
AIアシスト創薬:タンパク質結合剤のオールインワン自動設計
この記事は約13分で読めます。
最新のインシリコ創薬を支えるAI技術
近年、コンピュータ支援創薬(CADD)と 人工知能(AI)の統合が進んでいます。 AlphaFold によるタンパク質構造予測やタンパク質言語モデルといった革新的な技術の開発により、創薬プロセスの加速と個別化医療の進展が期待されています[1,2]。
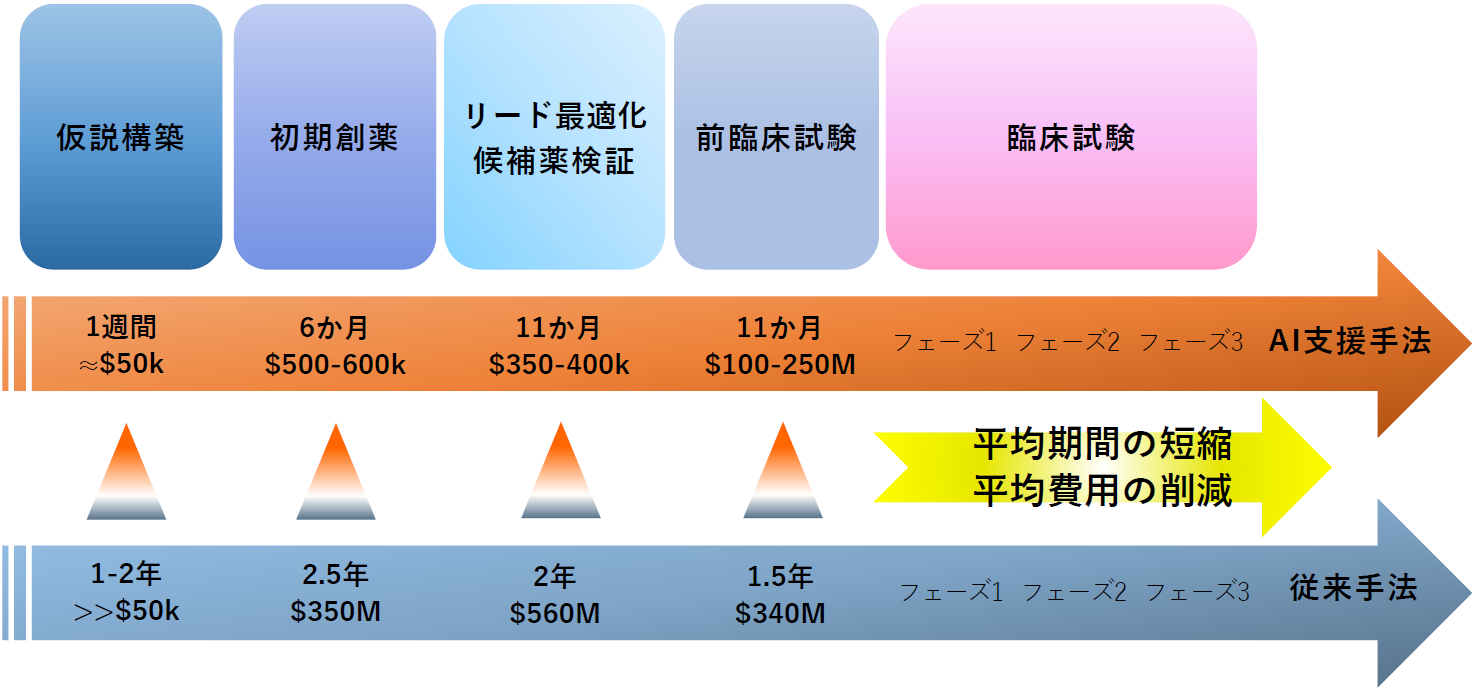
従来のハイスループットスクリーニング(HTS)では、試験できる化合物は通常数十万種類にとどまりますが、 AIを活用したバーチャルスクリーニング(VS)では、数百万種類もの化合物をはるかに高速に評価できます[3]。 また、機械学習(ML)や 深層学習(DL)に基づく生成モデルを用いるAIは、 既知の分子特性を分析し、最適な新規化学構造の設計を可能にします[4-6]。 AIは創薬プロセスの広いフェーズ(例えば、ヒット化合物・リード化合物の迅速な特定、医薬品分子構造設計の最適化、標的検証の加速など)に貢献しますが、 特に初期段階において、広大な化学空間を迅速かつ低コストで探索し、有望な新規候補物質を特定できる点は大きな強みです[4,5]。
さらに、AIの活用範囲は新薬開発にとどまりません。 患者個人の体質や病態に合わせて既存の治療法をカスタマイズする「個別化医療」への応用も進んでいます[5]。 このアプローチでは、患者の遺伝子情報や病気に関わるタンパク質に基づいて、治療効果の最大化と副作用の低減が図られます。 一律的な標準医療と比較すると、より安全で効率的な治療結果をもたらす事が重視され、がん治療などで普及が進んでいます[6]。
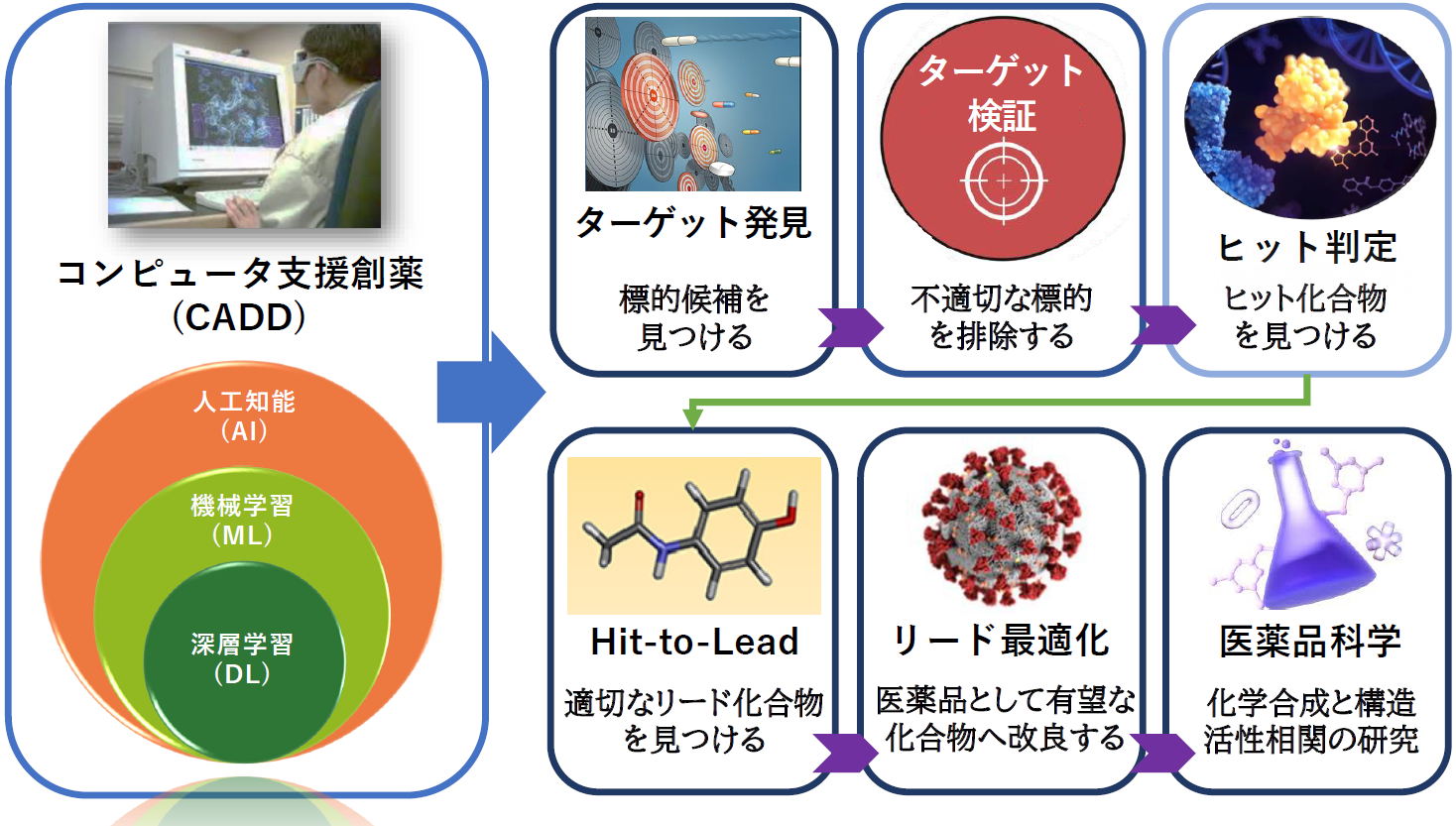
3DEXPERIENCE プラットフォーム上の BIOVIA Discovery Studio Simulation は、 機械学習や生成モデルを含むAI要素技術を、分子動力学(MD)シミュレーションを含む計算科学技術、 および独自に蓄積されたデータ資産と融合させて、タンパク質設計のための次世代ソリューションとして提供します。 強力なアルゴリズムを用いて複雑な創薬プロセスを最適化しますので、タンパク質設計における多様な課題に対応可能です。
適用例:
- 酵素活性部位の骨格設計
- 対称モチーフの骨格設計
- オリゴマータンパク質の設計
- タンパク質結合剤の設計


適用例: タンパク質結合剤の自動設計
結合剤設計の需要と課題
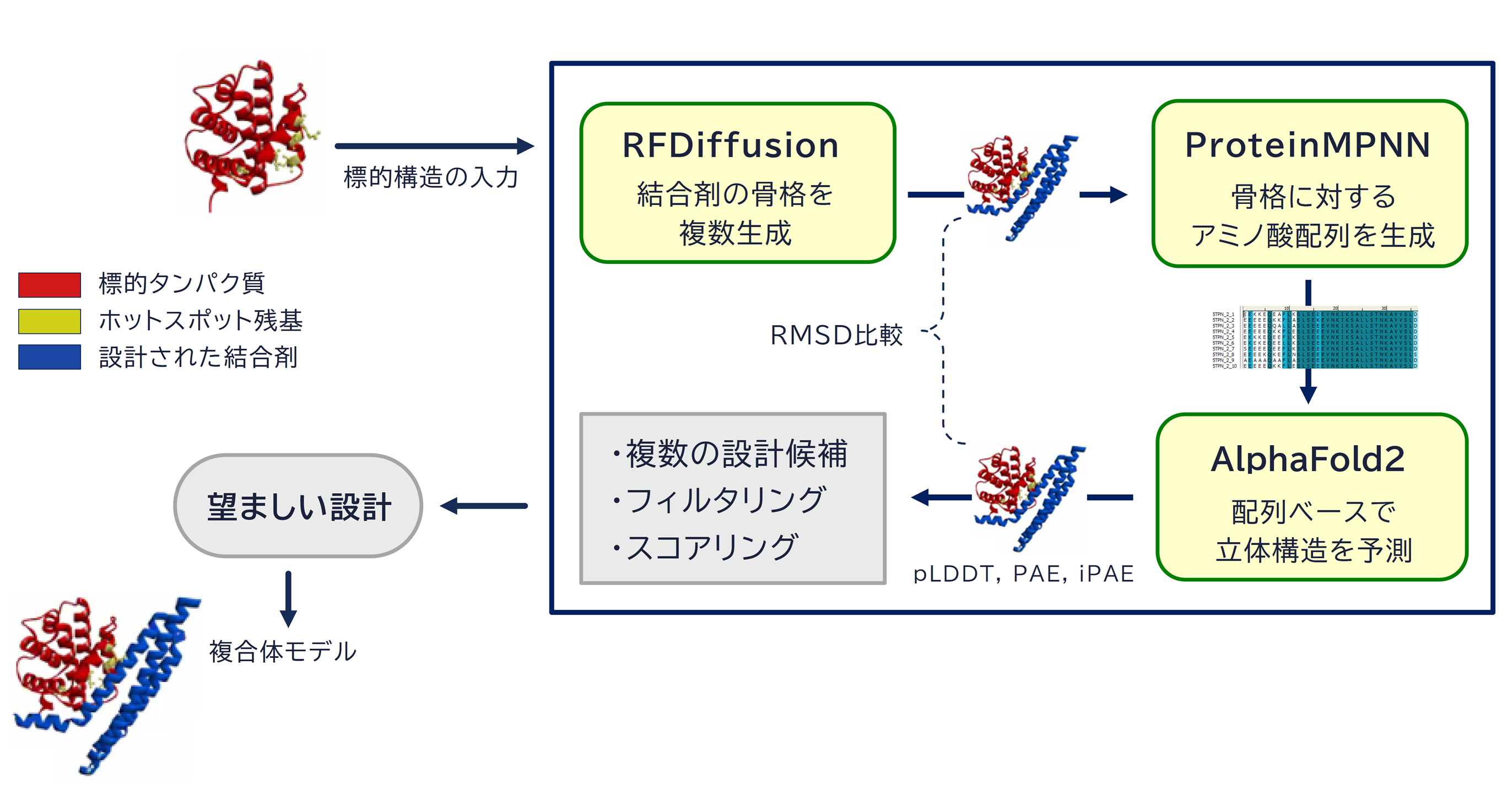
創薬、診断そして生命科学研究の現場において、高い親和性と特異性を持つタンパク質結合剤(Protein Binders)の存在は欠かせません。 タンパク質結合剤は、標的タンパク質を選択的に認識して結合できるタンパク質ベースのアフィニティー試薬であり、 特定の機能を持つタンパク質を発見または改良するために使用されています。 しかし、結合剤の設計は複雑であり、たとえ生成AIを用いたとしても容易ではありません。 生成AIによる設計アプローチの場合、一般的には次の複数のステップが必要となります[7,8]。
- 相互作用部位を考慮しながら、結合する新規分子の骨格候補を複数生成(RFdiffusion)[9]
- 新しい骨格に適合するアミノ酸配列候補を複数生成(ProteinMPNN)[10,11]
- 各配列に対して、望ましい結合が維持されるように構造が折り畳まれるか確認(AlphaFold2)[12]
設計全体を通して多数の設計パラメータ(生成条件や試行に関する可変パラメータ)を制御する必要があり、評価対象となる結合剤候補は膨大な数にのぼります[14]。 また厄介な事に、「あるプロジェクトでは成功したパラメータセットが、別の標的を用いたプロジェクトでも最適であるとは限らない」といった問題もあります。 このため、プロジェクトごとに広大なパラメータ空間を網羅的に探索できる、統合的な設計プロセスが求められます[15]。
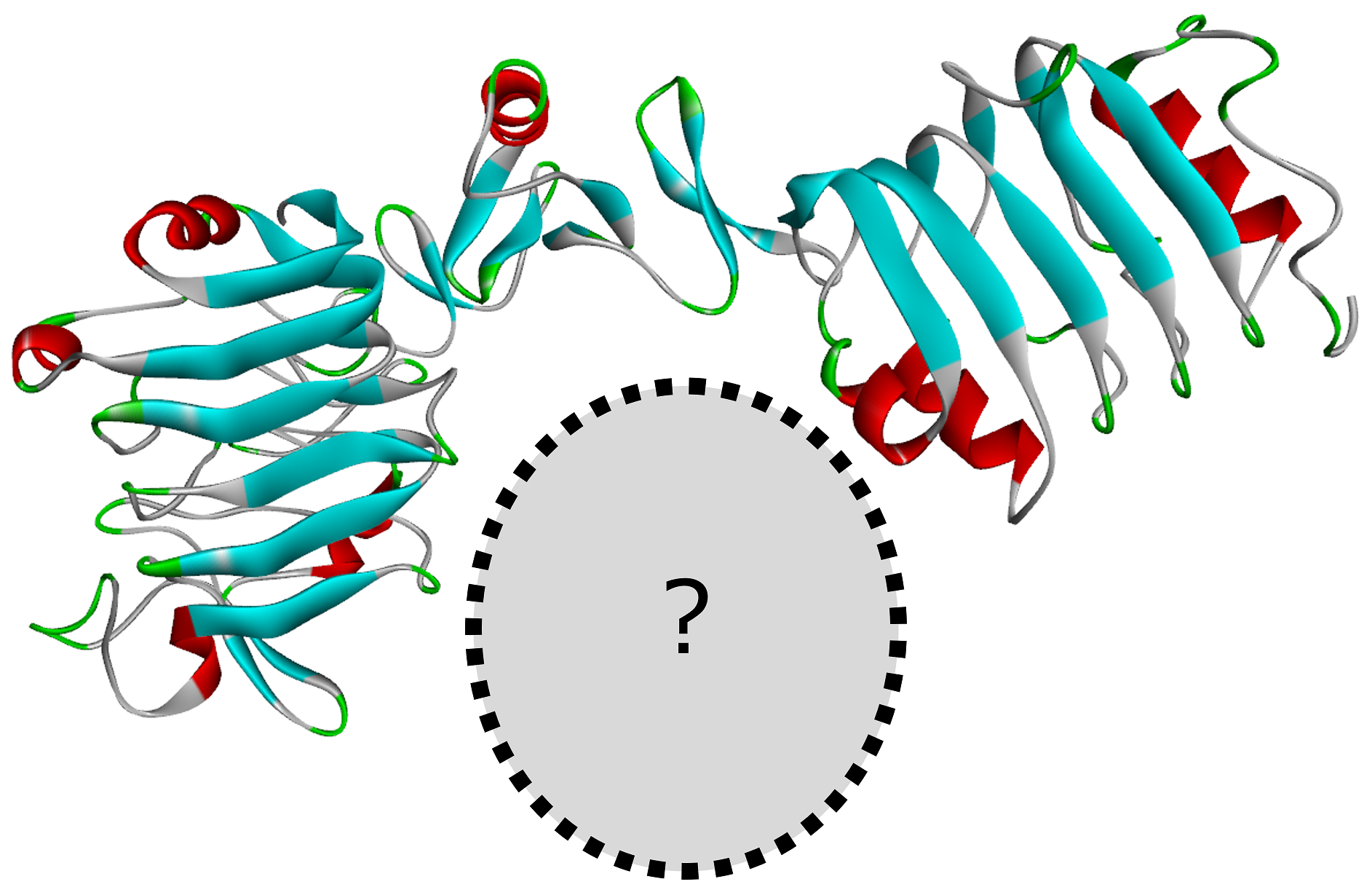
Design Protein Binders を用いた自動設計
上記のニーズに応えるツールが、 Discovery Studio Simulation R2025 で実装された Design Protein Binders です。 このツールの最大の特徴は、結合剤の構造と配列をオールインワンで自動設計可能 な点にあります。
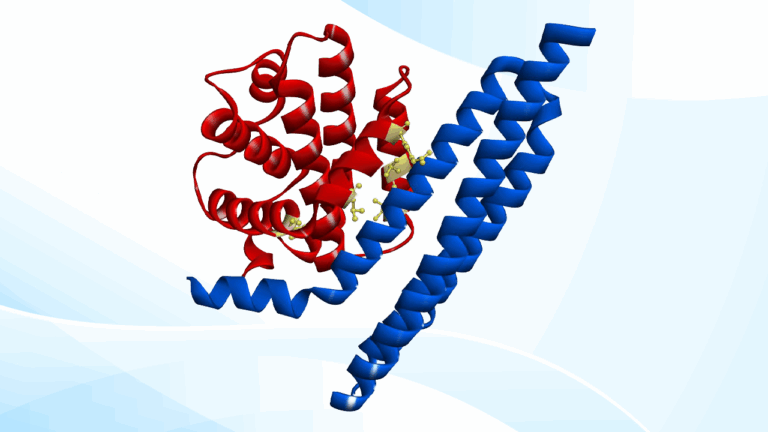
本ツールの実行手順は至って簡単です。
- 標的タンパク質の分子構造を用意します。PDBにあるデータをインポートして使用する事もできます。
- もし特定の相互作用部位(ホットスポット)の残基を標的としたい場合は、それらを指定する事ができます。
- 設計したい結合剤の大きさを、残基数の範囲で指定します。
- 実行ボタンを押すと、3DEXPERIENCE クラウド上のGPUリソースを利用して自動設計が開始されます。
この一連処理において、結合剤の構造と配列に関するデータはゼロから生成(デノボ設計)されます。 内部処理では RFdiffusion, ProteinMPNN, AlphaFold2 の各種生成AIが繰り返し呼び出され、 設計の最小要件を満たす結合剤候補が探索されます。 発見された候補はスコア関数によって評価され、更にスコアが良くなるようにパラメータセットが改良されていきます。 最終的に「望ましいパラメータセット」と「そこから得られる結合剤と標的の複合体構造」がスコアと共にリストアップされます。
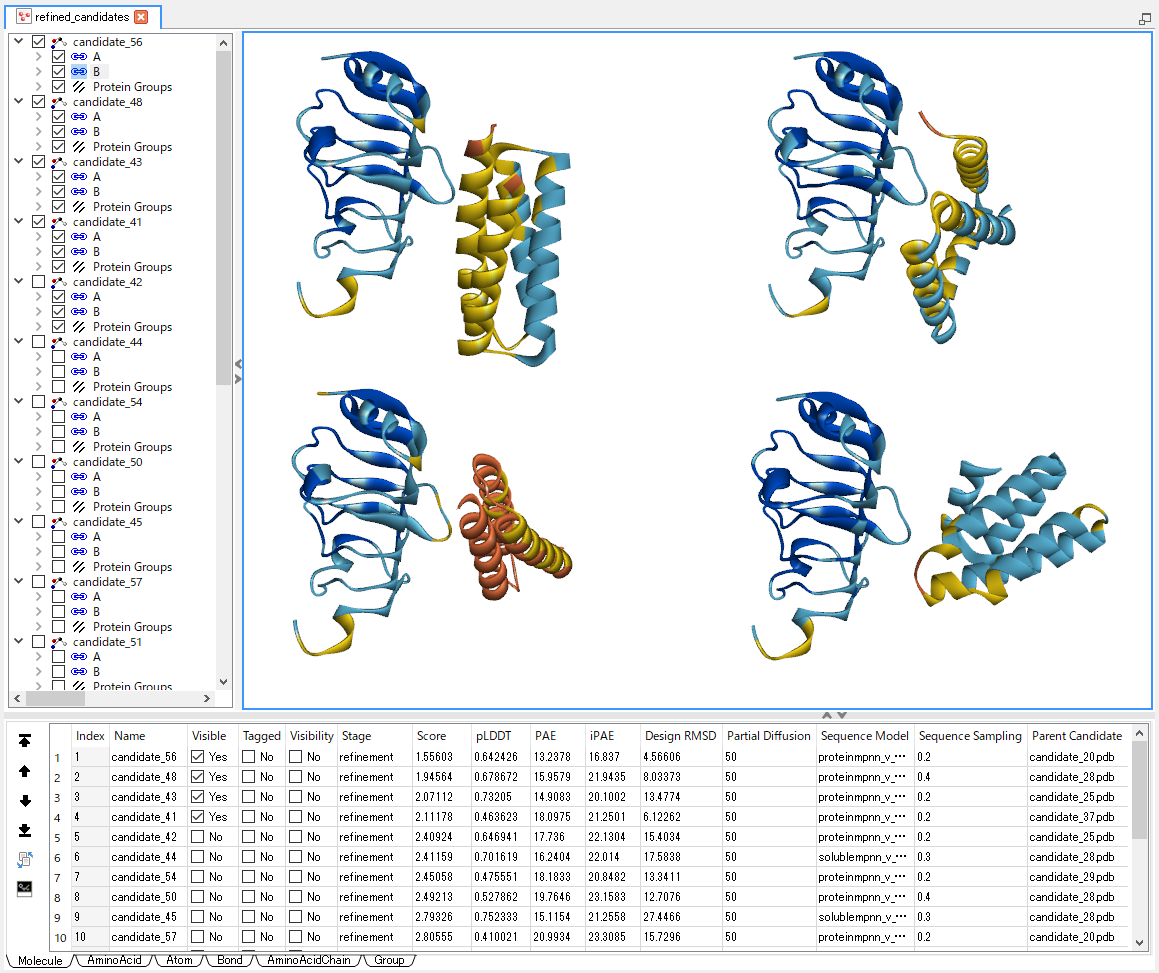
探索時および改良時におけるフィルタリング(最小設計要件)とスコアリング(設計の評価)には下記項目が使用されます。 各スコア項目について、フィルタ条件のカスタマイズや総合スコア計算式中の係数変更を行うことも可能です。
- pLDDT (局所構造の信頼度; AlphaFold2 算出)[12]
- PAE (ドメイン間相対位置の信頼度; AlphaFold2 算出)[12]
- iPAE (複合体結合面の信頼度)[9,13]
- RMSD (AlphaFold2 予測構造と RFdiffusion 予測構造の類似度)[9]
- NC末端距離 (構造のコンパクト性)
留意点:
本ツールは構造を直接的に最適化するのではなく、パラメータセットを最適化します。
結合剤の構造は、最適化されたパラメータセットから得られる出力となります。
手動設計ツールとの併用
前述のツールは、既存の生成ツールである、
- Predict Protein Structures ( AlphaFold2 )
- Generate Protein Scaffolds ( RFdiffusion )
- Generate Protein Sequences ( ProteinMPNN )
の3つを自動設計パイプラインへ統合したものとなります。 一方で、各生成ツールを個別に実行する事も従来通り可能です。 上位の結合剤候補を手動でさらに絞り込んだり、拘束条件を追加して再設計したり、 予測構造を再評価したりする場合に利用できます。

結合剤モデルの選定と追加評価
タンパク質結合剤のモデルが得られた後は、Discovery Studio でさらなる研究や設計へと進む事ができます。 精度の高い計算を行うために、モデル選定時に確認すべき事項を列挙します。
- 原子間の物理的接触
Discovery Studio のモニター機能では、原子同士の不自然な重なりが無いかを素早く確認できます。 側鎖衝突のような軽微な重なりであれば、簡易的なエネルギー最小化を実行する事で解消される場合があります。 - 分子間の引力相互作用
Analyze Protein Interface は、標的分子と結合分子との間の相互作用 (タンパク質間相互作用; Protein-Protein Interaction; PPI)を確認するために最適なツールです。 ツール実行前に水素原子の追加とエネルギー最小化を行っておくと、より現実に近い相互作用を評価する事ができます。 - 目的の分子特徴に関連する設計スコア
自動設計時のスコアは、得られたモデルの特徴ごとの信頼度に関わります。 例えば、相互作用部位の残基構造を正確に扱いたい場合は、iPAE は重要な指標の1つになります[9,13]。 もし球状タンパク質を優先して設計したい場合は、NC末端距離に代表されるような近接性も求められるでしょう。
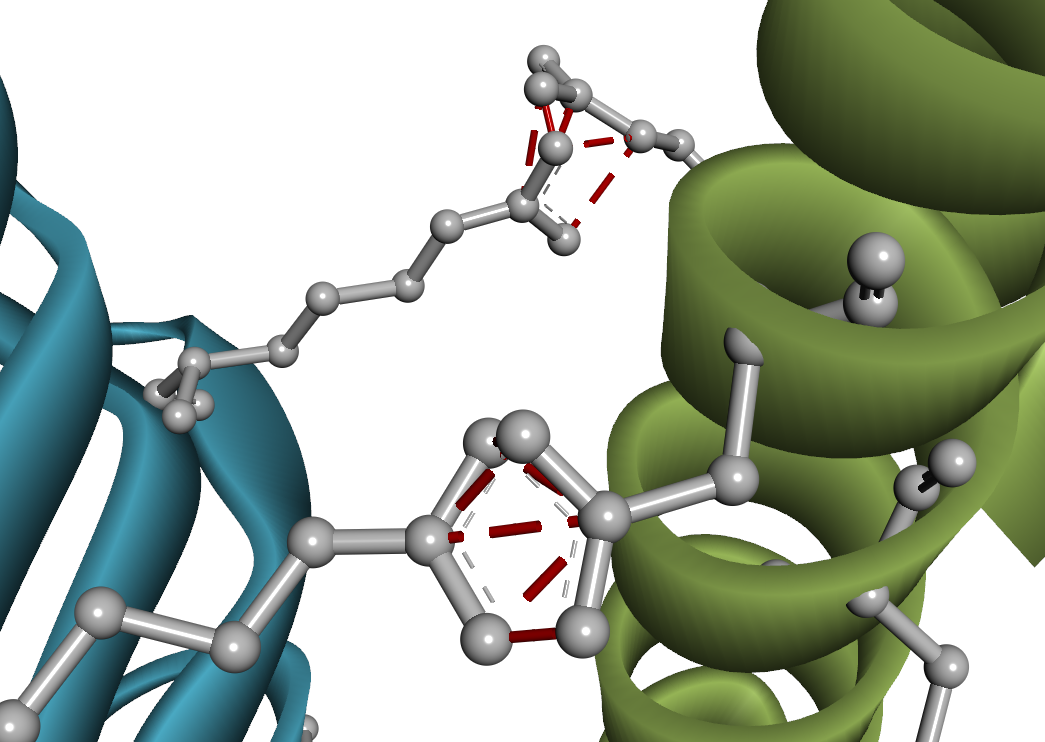
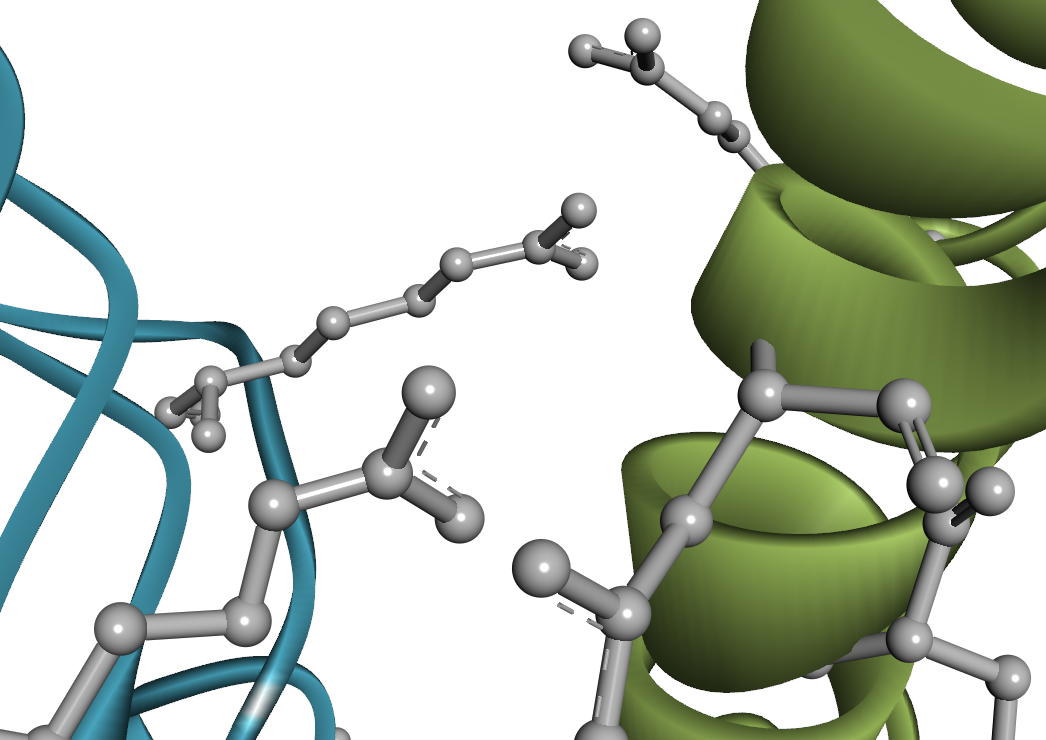
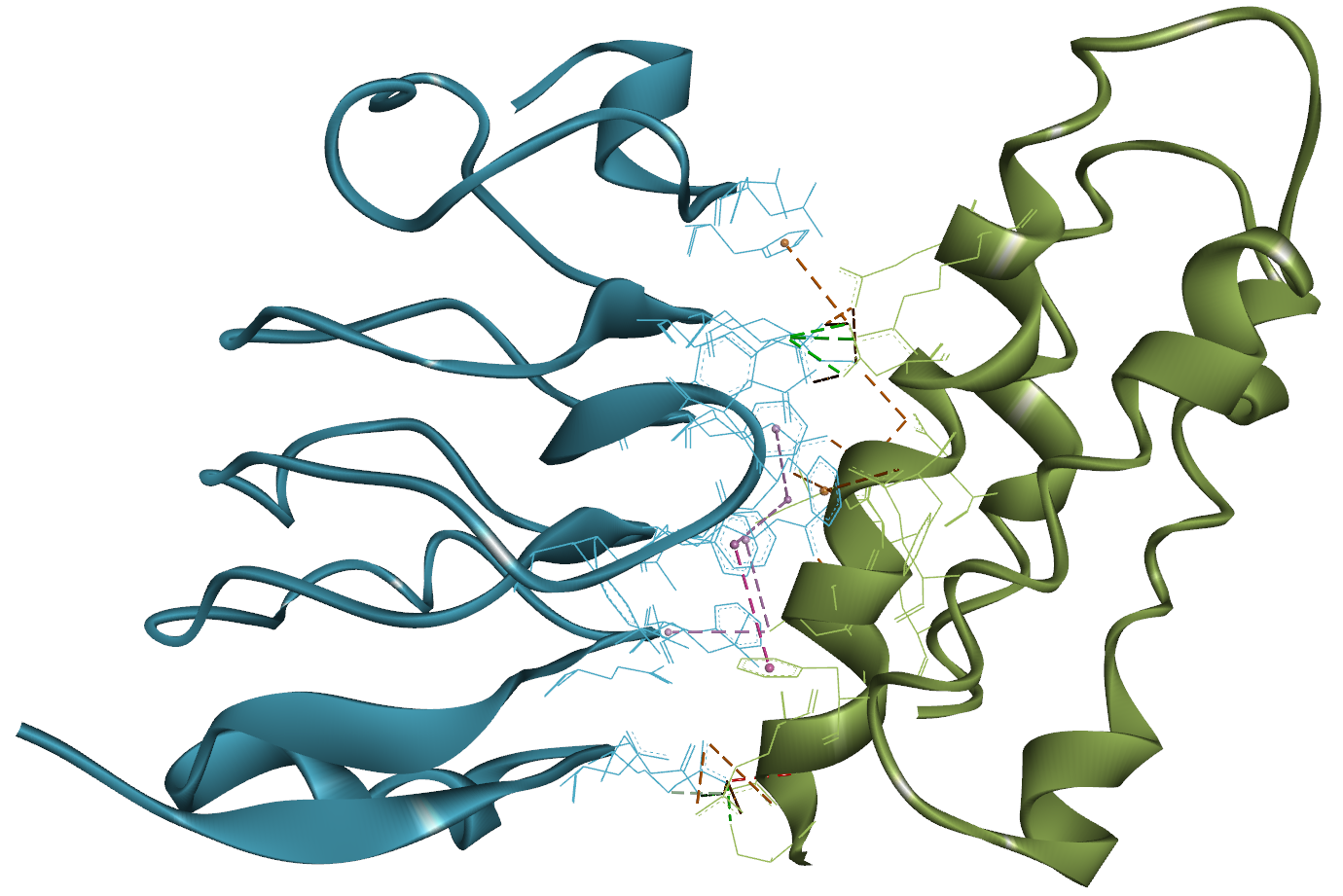
タンパク質結合剤の開発においては、 溶液中における遊離状態(単量体)と、標的タンパク質との結合状態(複合体)、両方の構造安定性も考慮しなければいけません。 Predict Protein Structures ツールでは AlphaFold2 を用いて、これら両方の状態を評価できます。 AlphaFold2 は Design Protein Binders ツール内部でも実行されていますが、 そちらでは単一配列の情報のみを用いて構造予測されるため、出力構造の精度が低下している可能性があります。 一方で Predict Protein Structures ツールでは、多重配列アラインメント(MSA)も考慮した再評価がなされます[19-21]。
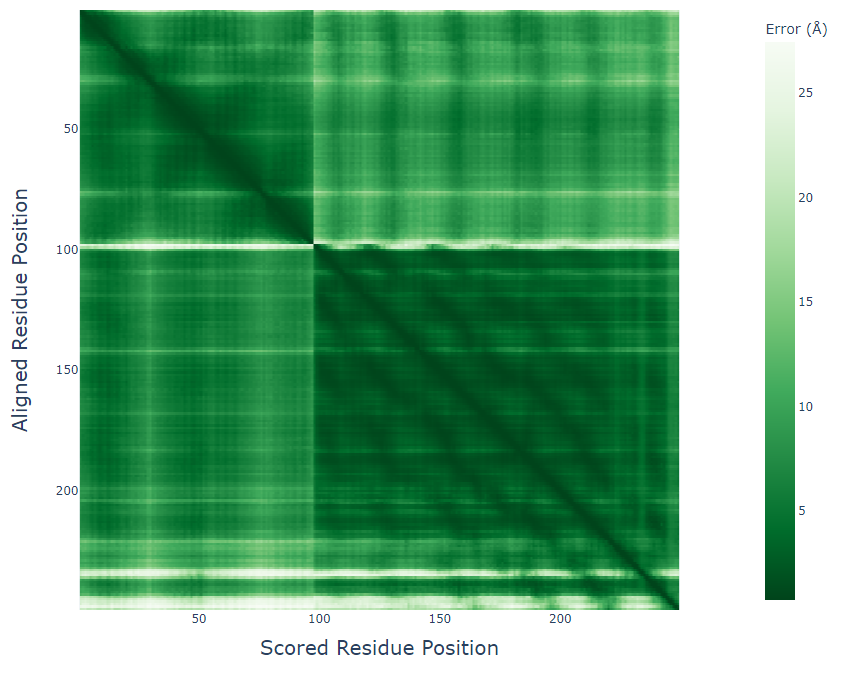
更なる安定性の検証として、MDシミュレーションを使用して、 設計された複合体の分子間相互作用が時間経過とともに維持されるかを調べることもできます。 MDシミュレーションに関しては記事を改めてご紹介したいと思います。
まとめ
本記事では、AIアシスト創薬の概説と BIOVIA Discovery Studio Simulation の新機能である Design Protein Binders を用いたタンパク質結合剤の自動設計手法を紹介しました。 記事内で紹介されたAIツールの他にも、多くのAIツールが今後追加されていく予定です。 もし本製品にご興味がございましたら、弊社ウェーブフロントまで お問い合わせ ください。
関連記事
立体構造構造決定の概説とDiscovery Studioを用いたホモロジーモデリング・AI支援型モデリングの紹介です。

参考文献
Trends Report by Discipline (2026), Life Science and Clinical Research Field, Center for Research and Development Strategy, Japan Science and Technology Agency, CRDS-FR-L105-202512, DOI:10.82643/crds-fr-l-hmc-aidd
Divya Vemula, et al. (2023), Eur. J. Pharm. Sci., 181(1): 106324, DOI:10.1016/j.ejps.2022.106324
H. Chen, D. Lu, Z. Xiao, et al. (2024), Health Inf. Sci. Syst., 12(1): 41, DOI:10.1007/s13755-024-00300-y
S. Ali, et al. (2025), J. Med. Chem., 68(22): 23643-23652, DOI:10.1021/acs.jmedchem.5c03159
Z. J. Baum, et al. (2021), J. Chem. Inf. Model., 61(7): 3197-3212, DOI:10.1021/acs.jcim.1c00619
V. Malheiro, et al. (2025), Pharmaceuticals, 18(6): 788, DOI:10.3390/ph18060788
LabCode (2025), "In-silico protein design for beginners using various tools: From de novo design to characterization.", Zenn, URL:zenn.dev/labcode/books/71b01f2557419e
N. R. Bennett, et al. (2023), Nat. Commun., 14(1): 2625, DOI:10.1038/s41467-023-38328-5
J. L. Watson, D. Juergens, N. R. Bennett, et al. (2023), Nature, 620(7976): 1089-1100, DOI:10.1038/s41586-023-06415-8
J. Dauparas, et al. (2022), Science, 378(6615): 49-56, DOI:10.1126/science.add2187
J. Dauparas, G. R. Lee, R. Pecoraro, et al. (2025), Nat. Methods, 22(4): 717-723, DOI:10.1038/s41592-025-02626-1
J. Jumper, R. Evans, A. Pritzel, et al. (2021), Nature, 596(7873): 583-589, DOI:10.1038/s41586-021-03819-2
R. Evans, M. O'Neill, A. Pritzel, et al. (2021), preprint: bioRxiv, DOI:10.1101/2021.10.04.463034
N. Gonzalez-Rodriguez, et al. (2025), PLOS Comput. Biol., 21(11): e1013747 DOI:10.1371/journal.pcbi.1013747
A. Nallathambi and B. Kuhlman (2025), Current Protocols, 5(10): e70208, DOI:10.1002/cpz1.70208
C. Chothia and J. Janin (1975), Nature, 256(5520): 705-708, DOI:10.1038/256705a0
A. R. Fersht, J. P. Shi, J. Knill-Jones, et al. (1985), Nature, 314(6008): 235-238, DOI:10.1038/314235a0
S. Jones and J. M. Thornton (1996), Proc. Natl. Acad. Sci. USA, 93(1): 13-20, DOI:10.1073/pnas.93.1.13
P. Bryant, G. Pozzati, and A. Elofsson (2022), Nat. Commun., 13(1): 1265 DOI:10.1038/s41467-022-28865-w
G. Ahdritz, N. Bouatta, C. Floristean, et al. (2024), Nat. Methods., 21(8): 1514-1524, DOI:10.1038/s41592-024-02272-z
Online tutorial, AlphaFold practical guide, EMBL-EBI Training, URL:ebi.ac.uk/training/online/courses/alphafold
K. M. Ruff and R. V. Pappu (2021), J. Mol. Biol., 433(20): 167208, DOI:10.1016/j.jmb.2021.167208
記事制作者
- 執筆:
- ララサティ・マルタ (株式会社ウェーブフロント 連成問題研究部)
- 佐橋 一裕 (株式会社ウェーブフロント 連成問題研究部)
- 編集:
- 佐橋 一裕 (株式会社ウェーブフロント 連成問題研究部)